1D Hubbard model#
Background physics#
To show the basic functionality of the package, we consider a simple interacting electronic system: a 1D chain of sites that allow nearest-neighbor tunneling with strength \(t\) and on-site repulsion \(U\) between two electrons if they are on the same site. Such a model is known as the 1D Hubbard model and is useful for understanding the onset of insulating phases in interacting metals.
To begin, we first consider the second quantized form of the non-interacting Hamiltonian. Because we expect the interacting ground state to be antiferromagnetic, we build a two-atom cell and name the two sublattices \(A\) and \(B\). These sublattices are identical to each other in the non-interacting case \(U=0\). The non-interacting Hamiltonian reads:
where \(\textrm{h.c}\) is the hermitian conjugate, \(\sigma\) denotes spin (\(\uparrow\) or \(\downarrow\)) and \(c_{i, A, \sigma}^{\dagger}\) creates an electron with spin \(\sigma\) in unit cell \(i\) of sublattice \(A\). Next up, is the interacting part of the Hamiltonian:
where \(n_{i, A, \sigma} = c_{i, A, \sigma}^{\dagger}c_{i, A, \sigma}\) is the number operator for sublattice \(A\) and spin \(\sigma\). The total Hamiltonian is then \(\hat{H} = \hat{H_0} + \hat{V}\). With the model defined, we can now proceed to input the Hamiltonian into the package and solve it using the mean-field approximation.
Problem definition#
Non-interacting Hamiltonian#
First, let’s get the basic imports out of the way.
import numpy as np
import matplotlib.pyplot as plt
import meanfi
Now let us translate the non-interacting Hamiltonian \(\hat{H_0}\) defined above into the basic input format for the package: a tight-binding dictionary.
The tight-binding dictionary is a python dictionary where the keys are tuples of integers representing the hopping vectors and the values are the hopping matrices.
For example, a key (0,) represents the onsite term in one dimension and a key (1,) represents the hopping a single unit cell to the right.
In two dimensions a key (0,0) would represent the onsite term and (1,0) would represent hopping to the right in the direction of the first reciprocal lattice vector.
In the case of our 1D Hubbard model, we only have an onsite term and hopping a single unit cell to the left and right.
Thus our non-interacting Hamiltonian becomes:
hopp = np.kron(np.array([[0, 1], [0, 0]]), np.eye(2))
h_0 = {(0,): hopp + hopp.T.conj(), (1,): hopp, (-1,): hopp.T.conj()}
Here hopp is the hopping matrix which we define as a kronecker product between sublattice and spin degrees of freedom: np.array([[0, 1], [0, 0]]) corresponds to the hopping between sublattices and np.eye(2) leaves the spin degrees of freedom unchanged.
In the corresponding tight-binding dictionary h_0, the key (0,) contains hopping within the unit cell and the keys (1,) and (-1,) correspond to the hopping between the unit cells to the right and left respectively.
To verify the validity of h_0, we evaluate it in the reciprocal space using the tb_to_kgrid, then diagonalize it and plot the band structure:
nk = 50 # number of k-points
ks = np.linspace(0, 2 * np.pi, nk, endpoint=False)
hamiltonians_0 = meanfi.tb_to_kgrid(h_0, nk)
vals, vecs = np.linalg.eigh(hamiltonians_0)
plt.plot(ks, vals, c="k")
plt.xticks([0, np.pi, 2 * np.pi], ["$0$", "$\pi$", "$2\pi$"])
plt.xlim(0, 2 * np.pi)
plt.ylabel("$E - E_F$")
plt.xlabel("$k / a$")
plt.show()
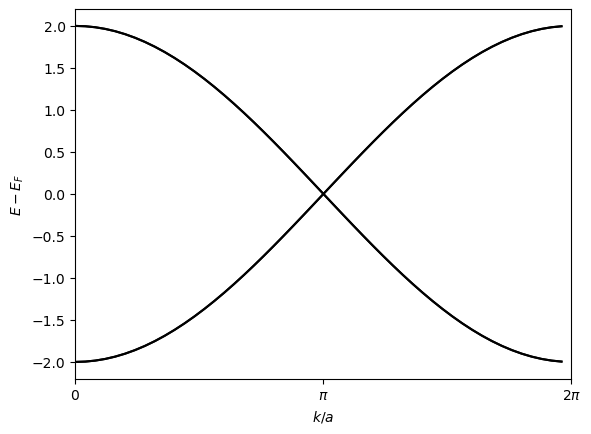
which seems metallic as expected.
Interaction Hamiltonian#
We now proceed to define the interaction Hamiltonian \(\hat{V}\).
To achieve this, we utilize the same tight-binding dictionary format as before.
Because the interaction Hamiltonian is on-site, it must be defined only for the key (0,) and only for electrons on the same sublattice with opposite spins.
Based on the kronecker product structure we defined earlier, the interaction Hamiltonian is:
U = 2
s_x = np.array([[0, 1], [1, 0]])
h_int = {
(0,): U * np.kron(np.eye(2), s_x),
}
Here s_x is the Pauli matrix acting on the spin degrees of freedom, which ensures that the interaction is only between electrons with opposite spins whereas the np.eye(2) ensures that the interaction is only between electrons on the same sublattice.
Putting it all together#
To combine the non-interacting and interaction Hamiltonians, we use the Model class.
In addition to the Hamiltonians, we also need to specify the filling of the system — the number of electrons per unit cell.
filling = 2
full_model = meanfi.Model(h_0, h_int, filling)
The object full_model now contains all the information needed to solve the mean-field problem.
Solving the mean-field problem#
To find a mean-field solution, we first require a starting guess.
In cases where the non-interacting Hamiltonian is highly degenerate, there exists several possible mean-field solutions, many of which are local and not global minima of the energy landscape.
Therefore, the choice of the initial guess can significantly affect the final solution depending on the energy landscape.
Here the problem is simple enough that we can generate a random guess for the mean-field solution through the guess_tb function.
It creates a random Hermitian tight-binding dictionary based on the hopping keys provided and the number of degrees of freedom within the unit cell.
Because the mean-field solution cannot contain hoppings longer than the interaction itself, we use h_int keys as an input to guess_tb.
Finally, to solve the model, we use the solver function which by default employes a root-finding algorithm to find a self-consistent mean-field solution.
filling = 2
full_model = meanfi.Model(h_0, h_int, filling)
guess = meanfi.guess_tb(frozenset(h_int), ndof=4)
mf_sol = meanfi.solver(full_model, guess, nk=nk)
The solver function returns only the mean-field correction to the non-interacting Hamiltonian in the same tight-binding dictionary format.
To get the full Hamiltonian, we add the mean-field correction to the non-interacting Hamiltonian and plot the band structure just as before:
h_mf = meanfi.add_tb(h_0, mf_sol)
hamiltonians = meanfi.tb_to_kgrid(h_mf, nk)
vals, vecs = np.linalg.eigh(hamiltonians)
plt.plot(ks, vals, c="k")
plt.xticks([0, np.pi, 2 * np.pi], ["$0$", "$\pi$", "$2\pi$"])
plt.xlim(0, 2 * np.pi)
plt.ylabel("$E - E_F$")
plt.xlabel("$k / a$")
plt.show()
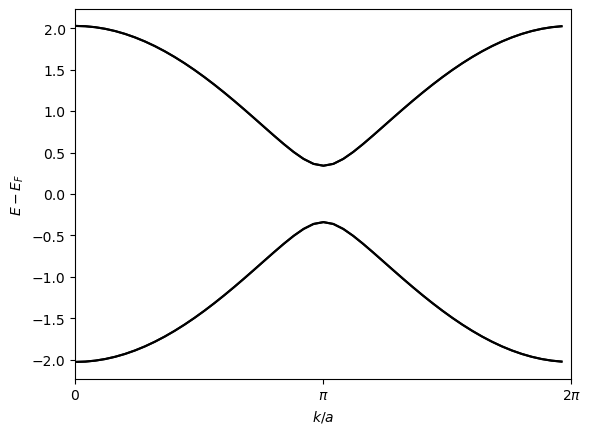
the band structure now shows a gap at the Fermi level, indicating that the system is in an insulating phase!
We can go further and compute the gap for a wider range of \(U\) values:
def compute_sol(U, h_0, nk, filling=2):
h_int = {
(0,): U * np.kron(np.eye(2), np.ones((2, 2))),
}
guess = meanfi.guess_tb(frozenset(h_int), len(list(h_0.values())[0]))
full_model = meanfi.Model(h_0, h_int, filling)
mf_sol = meanfi.solver(full_model, guess, nk=nk)
return meanfi.add_tb(h_0, mf_sol)
def compute_gap(full_sol, nk_dense, fermi_energy=0):
h_kgrid = meanfi.tb_to_kgrid(full_sol, nk_dense)
vals = np.linalg.eigvalsh(h_kgrid)
emax = np.max(vals[vals <= fermi_energy])
emin = np.min(vals[vals > fermi_energy])
return np.abs(emin - emax)
def compute_phase_diagram(
Us,
nk,
nk_dense,
):
gaps = []
for U in Us:
full_sol = compute_sol(U, h_0, nk)
gaps.append(compute_gap(full_sol, nk_dense))
return np.asarray(gaps, dtype=float)
Us = np.linspace(0, 4, 30, endpoint=True)
gaps = compute_phase_diagram(Us=Us, nk=20, nk_dense=100)
plt.plot(Us, gaps, c="k")
plt.xlabel("$U / t$")
plt.ylabel("$\Delta{E}/t$")
plt.show()
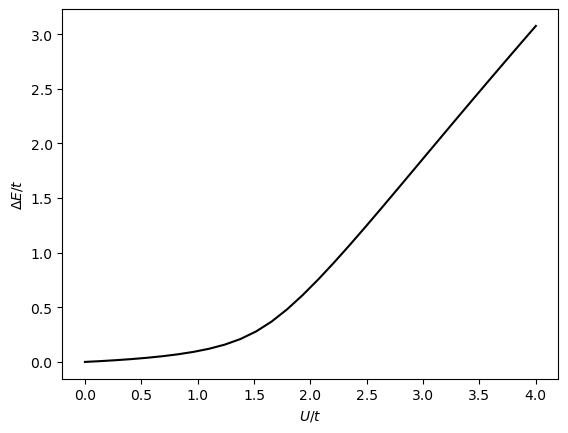
We see that at around \(U=1\) the gap opens up and the system transitions from a metal to an insulator. In order to more accurately determine the size of the gap, we chose to use a denser k-grid for the diagonalization of the mean-field solution.